A Dockerized PacBio Workflow that Slashed Run-Time by 41%
- sdshinghal
- Nov 28, 2024
- 2 min read

Note: This is a republication of a blog post I wrote for Strand Life Sciences, published on 28/11/2024. The original can be found here.
Run time can become a major bottleneck in analysis when working with large file sizes. So, when approached by a client to build a long-read analysis pipeline, we created a dockerized solution that cut down run time by 41%. Our pipeline produced a vcf containing over 50,000 variants from a 42 GB file in just 120 minutes. When run through the various tools individually, this same analysis took 204 minutes.
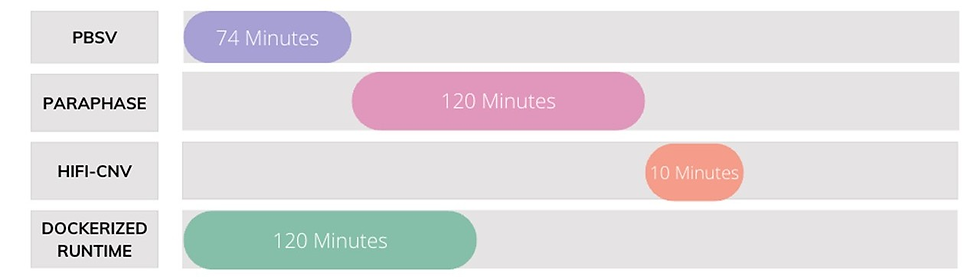
The client was working with unaligned long-reads obtained from a Pacific Biosciences machine, however, the analysis proved tedious and time-consuming. Each tool required separate setup, configuration, and resource allocation, leading to inconsistent environments, inefficiency in execution, and fragmented outputs.
We created a unified Docker-based solution that packaged these scattered workflows into one whole and optimized them such that it would be -$1.24 cheaper to run a typical 100GB WGS using this solution, and when run for one month, you would be able to run 62 more WGS samples as compared to running them through each tool separately.

Through this Docker solution, we were able to:
Manage different dependencies for each tool within a single Docker image without version conflicts
Ensure tools can run in parallel without interference while handling interdependencies when they exist
Implement standardized logging and robust error handling
Ensure the Docker image is flexible for additional tools and scalable for larger datasets in the future
Minimize Docker overhead and improve runtime efficiency to prevent the image from slowing down the analysis pipeline.
Store parameters in config files for easy sharing and reproducibility
Provide the option to run a subset of tools as per user’s need.

Comments